Desde Duchenne y tú, nos unimos un año más a la conmemoración del Día Nacional de las Enfermedades Neuromusculares, una iniciativa impulsada por la Federación Española de Enfermedades Neuromusculares (ASEM) que se celebra el 15 de noviembre.
En este año 2023, la fecha coincide con el vigésimo aniversario de la constitución de la Federación ASEM. El objetivo de este día sigue siendo la concienciación y difusión del conocimiento de estas enfermedades y seguir visibilizando la lucha por la inclusión social, la accesibilidad a los tratamientos y la mejora de la calidad de vida de los pacientes y sus familias.
Las enfermedades neuromusculares constituyen un grupo diverso de más de 150 trastornos que afectan al Sistema Nervioso Periférico y que se diferencian y clasifican por la localización del daño:1
- Enfermedades que afectan a las neuronas motoras cuya función es producir estímulos para la contracción de grupos musculares implicados en la realización de actividades como caminar, hablar, gesticular, tragar, etc.
- Neuropatías en las que están afectados los troncos nerviosos y nervios periféricos que dan lugar a dolor, adormecimiento, cosquilleo y debilidad muscular en distintas partes del cuerpo.
- Miopatías y distrofias musculares, en estos casos están afectados los músculos.
- Miastenias, en las que está afectada la comunicación entre el nervio y el músculo, caracterizadas por debilidad muscular variable que aparece tras la realización de actividad física y que se recupera con el reposo.
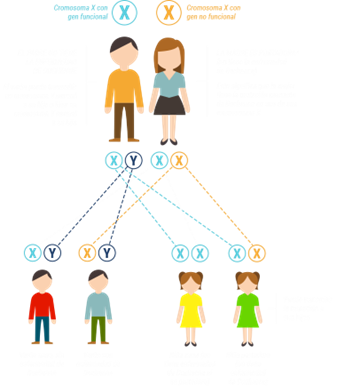
La mayoría de estas enfermedades son de origen genético, como las miopatías congénitas, la distrofia muscular de Duchenne y la atrofia muscular espinal, y, por lo tanto, pueden ser hereditarias y trasmisibles a la descendencia. Sin embargo, también pueden ser el resultado de una respuesta inmunitaria anormal, como la miastenia gravis, de infecciones o exposiciones a tóxinas.2
La aparición de las enfermedades neuromusculares puede producirse en cualquier etapa de la vida, pero más del 50% debutan en la infancia.3 La mayoría de las que debutan en la infancia tienen una base genética. La afección neuromuscular genética pediátrica más común es la distrofia muscular de Duchenne, que afecta a 1 de cada 3.600 a 6.000 varones recién nacidos en todo el mundo.4
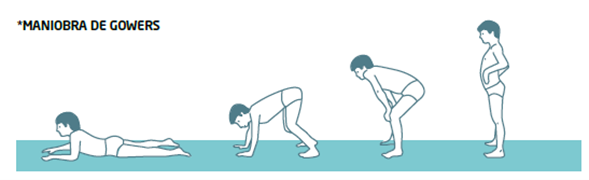
Aunque los síntomas pueden variar de una patología a otra, el síntoma fundamental de las enfermedades neuromusculares es la pérdida progresiva de fuerza muscular y la degeneración del conjunto de los músculos y nervios que los controlan.5
Un diagnóstico temprano puede facilitar el acceso al tratamiento y a los servicios adecuados, ya que el manejo clínico de estas patologías requiere un abordaje multidisciplinar con la participación de diversos especialistas (neurólogo, neuropediatra, rehabilitador, neumólogo, cardiólogo, cirujano ortopeda, endocrino, nutricionista, médico de atención primaria, fisioterapeuta, terapeuta ocupacional) que contribuyen a mejorar la evolución de los pacientes y retrasar así las posibles complicaciones.3-5
Aunque no existen tratamientos curativos para la mayoría de las enfermedades neuromusculares, existe la necesidad de seguir investigando en diversas áreas:1
- Desarrollo de nuevas herramientas de diagnóstico genético más avanzadas para lograr un diagnóstico temprano.
- Identificación de biomarcadores que faciliten la identificación precoz de estos trastornos, así como su seguimiento y evaluación de intervenciones terapéuticas.
- Desarrollo de terapias de precisión que buscan corregir los defectos genéticos subyacentes de estas enfermedades neuromusculares.
Bibliografía:
- Hospital Universitario 12 de Octubre. Enfermedad Neuromuscular. Disponible en: https://www.comunidad.madrid/hospital/12octubre/profesionales/servicios-medicos/enfermedad-neuromuscular
- AEPED. En familia. Enfermedades Neuromusculares. Disponible en: https://enfamilia.aeped.es/temas-salud/enfermedades-neuromusculares
- ASEM. ¿Qué son las enfermedades neuromusculares? Disponible en: https://www.asem-esp.org/que-son/
- PTC campus. ¿Qué son las distrofias musculares? Disponible en: https://ptccampus.es/dmd
- ASEM. Guía de las Enfermedad Neuromusculares. Información y apoyo a las familias. Disponible en: http://www.asemgalicia.com/wp-content/uploads/Guia-enfermedades-neuromusculares-informacion-apoyo-familias.pdf